Human and Monkey Chromosomes
Human chromosome 2 fusion (46 vs 48 in apes) marks hominin divergence ~3-4 Mya. Neanderthals/Denisovans shared this fusion. Homo erectus survived 2M years. Genomics reveals 7% uniquely human DNA, enriched for brain development. Human Accelerated Regions (HARs) drove neocortex expansion via altered RNA folding in the ventricular zone. Genes ARHGAP11B and NOTCH2NL triggered cortical folding through outer radial glia expansion. ATAC-seq and brain organoids allow causal testing of mutations. Human cognition emerges from lawful, testable genomic protocols—value flows from individual verification, not imposed doctrine. The 3M-year gap between genetic change and behavioral modernity remains the open mystery.
Chromosomes in Humans vs. Monkeys
Humans have 46 chromosomes, organized in 23 pairs — 22 pairs of autosomes plus 1 pair of sex chromosomes (XX or XY).
“Monkeys” is actually a broad category, so the number varies by species:
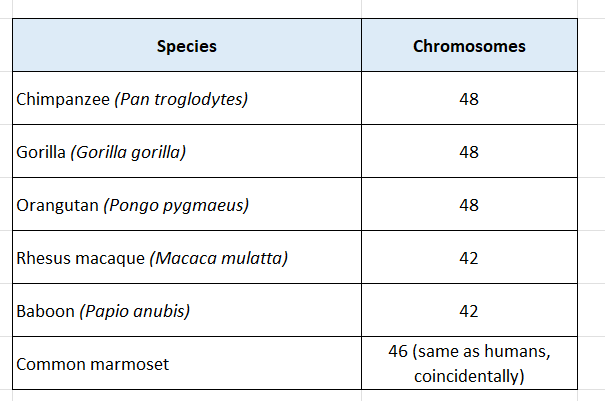
The Fascinating Discrepancy
The fact that humans have 2 fewer chromosomes than our closest relatives (chimps, gorillas, orangutans) is one of the most elegant pieces of evolutionary evidence. Human chromosome 2 is widely understood to be the result of a fusion of two ancestral chromosomes — it still carries vestigial telomeric sequences right in its middle, and a remnant of a second centromere, which are the “scars” of that ancient fusion event.
This is a cornerstone finding in comparative genomics and directly relevant to human DNA databases, since chromosome 2 is one of the most studied in terms of our divergence from other great apes.
When Did the Chromosome Fusion Happen?
The short answer: roughly 3 to 4 million years ago, after the human lineage split from the chimpanzee lineage.
The Timeline
~6–7 million years ago — The common ancestor of humans and chimpanzees diverges. At this point, the ancestor likely still had 48 chromosomes.
~3–4 million years ago — The fusion event is estimated to have occurred, somewhere in the early hominin lineage (possibly around Australopithecus or earlier).
By the time of Homo sapiens (~300,000 years ago), the 46-chromosome karyotype was already long established and universal in our lineage.
How Do We Know the Date?
The dating is inferred through molecular clock analysis — by comparing the mutation rates and divergence of the DNA sequences around the fusion site on chromosome 2 with equivalent regions in chimpanzee chromosomes 2a and 2b. The “scar” on human chromosome 2 is so well-preserved that it can be read almost like a fossil.
An Important Nuance
The fusion would have first appeared as a mutation in a single individual. That individual had 47 chromosomes — one fused, one unfused from each ancestral pair. Over generations, if there was no strong selective disadvantage (and there wasn’t), it spread through the population and eventually became fixed — meaning universal in our ancestral lineage.
This is actually a beautiful example of how a major chromosomal change can propagate through a population without necessarily causing immediate harm.
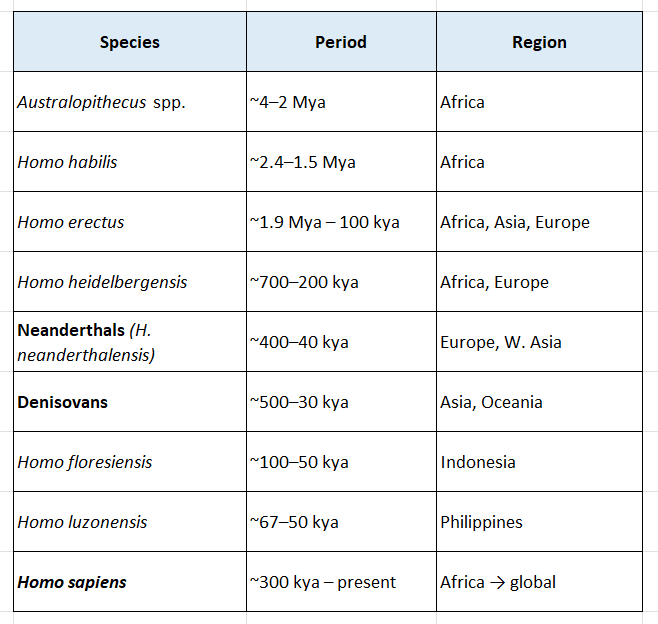
The Mosaic of Hominin Species and Interbreeding
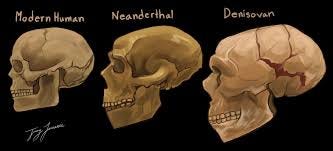
This is one of the most exciting frontiers in paleoanthropology and ancient DNA research. The picture is far more complex — and far more interesting — than a simple linear progression.
The Major Players
The Interbreeding Evidence — Ancient DNA Revolution
This is where genomics completely rewrote the textbooks, particularly after Svante Pääbo’s work (Nobel Prize 2022):
Modern non-African humans carry ~1–4% Neanderthal DNA — the interbreeding happened roughly 50,000–60,000 years ago as H. sapiens left Africa.
Melanesians and Aboriginal Australians carry ~4–6% Denisovan DNA — a separate interbreeding event in Asia/Oceania.
Some African populations show traces of interbreeding with as-yet-unidentified archaic hominins — a “ghost lineage” not yet found in the fossil record.
There is also evidence of Neanderthal-Denisovan interbreeding — they mixed with each other too, not just with us.
The “Ghost Lineages” Problem
This is philosophically fascinating: ancient DNA analysis has revealed populations we have never found fossils of. We know they existed only because their genetic signature lives on in modern humans. The genome becomes a kind of palimpsest — layers of ancient presences written into our cells.
What This Means for the Chromosome Question
The 46-chromosome fusion was likely already fixed well before all this interbreeding. Neanderthals and Denisovans also had 46 chromosomes — confirming the fusion predates the split between these lineages and H. sapiens, which occurred roughly 500,000–700,000 years ago.
So the fused chromosome 2 is something we share with Neanderthals and Denisovans — it’s one of the markers of the broader Homo clade, not exclusive to sapiens.
Homo erectus — The Great Survivor
The crucial point. Homo erectus is arguably the most successful hominin species that ever existed, measured purely by duration.
The Numbers Are Staggering
Appeared: ~1.9 million years ago
Last known survival: ~100,000–50,000 years ago (Ngandong, Java)
Total span: nearly 2 million years
For comparison, Homo sapiens has existed for only ~300,000 years. We are, in terms of longevity, still a very young species.
What Made H. erectus So Resilient?
First hominin to leave Africa — spreading across the Caucasus, India, Southeast Asia, China
Mastered fire (evidence from ~1 Mya at Wonderwerk Cave, South Africa)
Acheulean tool technology — a major cognitive leap
Larger brain than predecessors (~900cc vs ~650cc in habilis)
Body proportions close to modern humans — long legs, built for endurance walking
The Overlapping Coexistence Problem
This is where it gets deeply complex:

H. erectus at Ngandong (Java) may have been contemporaneous with modern humans already present in the region. Whether they met, and whether there was any gene flow, remains an open and burning question in paleoanthropology.
The “Hobbit” Connection
Homo floresiensis — the tiny-brained hominin of Flores island — is now thought by many researchers to be a dwarfed descendant of H. erectus, shaped by island evolution. If so, H. erectus as a lineage survived in isolated pockets potentially until ~50,000 years ago — possibly encountering the first modern humans to reach Oceania.
A Philosophical Note
There is something profound about H. erectus. It walked the Earth for nearly 2 million years across three continents, adapted to glacials and interglacials, coexisted with multiple other hominin species — and yet left no living descendants (or perhaps only traces through unknown interbreeding events).
It is the great silent presence in our deep past.
The Mysterious 7% — Uniquely Human DNA
This is a very real and fascinating finding, and you’re likely referring to research that made significant waves in paleogenomics around 2021.
The Core Finding
A study by Nathan Schaefer et al. (UC Santa Cruz, published in Science Advances, 2021) analyzed the genomes of modern humans, Neanderthals, and Denisovans and concluded that approximately 7% of the modern human genome is uniquely ours — meaning it is not shared with Neanderthals, Denisovans, or other archaic hominins.
This 7% represents the genetic territory that defines Homo sapiens as a distinct biological entity.
What Does That 7% Contain?
Intriguingly, the uniquely human regions are disproportionately associated with:
Brain development and neural connectivity — particularly genes involved in synapse formation and cortical organization
Wound healing and immune response
Skeletal and facial morphology — contributing to the distinctly gracile human skull
Spermatogenesis — reproductive biology
This suggests the 7% is not random drift — it carries functionally significant adaptations.
When Did It Accumulate?
This is the most intriguing part. It didn’t happen all at once — it accumulated over time, but with key acceleration windows:

So the 7% was gradually built, but some researchers argue there was a notable acceleration in the window between 300,000 and 50,000 years ago.
The Related Concept — Human Accelerated Regions (HARs)
Complementing the 7% finding, Katherine Pollard (UCSF) identified roughly 2,700 “Human Accelerated Regions” — short DNA segments that are nearly identical across all mammals but mutated far faster than normal in the human lineage specifically. The most famous, HAR1, is active during fetal brain development — particularly in the Cajal-Retzius neurons that organize the cerebral cortex.
This suggests the cognitive leap that made us human has a very specific, identifiable genomic signature.
The Open Mystery
What caused this acceleration? Hypotheses include:
A population bottleneck creating intense selective pressure
Biased gene conversion during recombination
A viral integration event — some researchers point to ancient retroviruses embedded in our genome that may have rewired gene regulation
Simple positive selection for intelligence and social complexity
The honest answer is: we don’t fully know yet. And that 7% may be one of the deepest biological definitions of what it means to be human.
Human Accelerated Regions (HARs) and Brain Evolution — A Dated Deep Dive

1. The Discovery of HARs
2006 — Katherine Pollard and colleagues published the landmark paper in Nature identifying 202 Human Accelerated Regions. The methodology was elegant: compare the genomes of humans, chimpanzees, and other vertebrates, and find segments that were conserved for hundreds of millions of years across mammals — but then suddenly mutated rapidly and specifically in the human lineage after our split from chimpanzees.
The logic is powerful: if something was conserved for 300 million years across fish, reptiles, and mammals, it was under extreme evolutionary constraint — meaning it was critically important and mutation was lethal. When it suddenly changed only in humans, something extraordinary happened.
2. HAR1 — The Crown Jewel
HAR1 is the most dramatic example:
It changed only 2 bases across 310 million years of vertebrate evolution
Then changed 18 bases in the ~6 million years since the human-chimp split
That is an acceleration rate of approximately 300x the background mutation rate
What does HAR1 do?
It encodes part of a non-coding RNA gene (HAR1F) active in a very specific time window:

The human neocortex has 6 distinct layers. This layering is the structural basis of complex cognition, language, and abstract thought. HAR1 appears to be one of the genetic switches that builds this architecture differently — and more elaborately — in humans than in any other primate.
3. The Timeline of Cortical Expansion
This is where genetics meets paleoanthropology with precise dates:
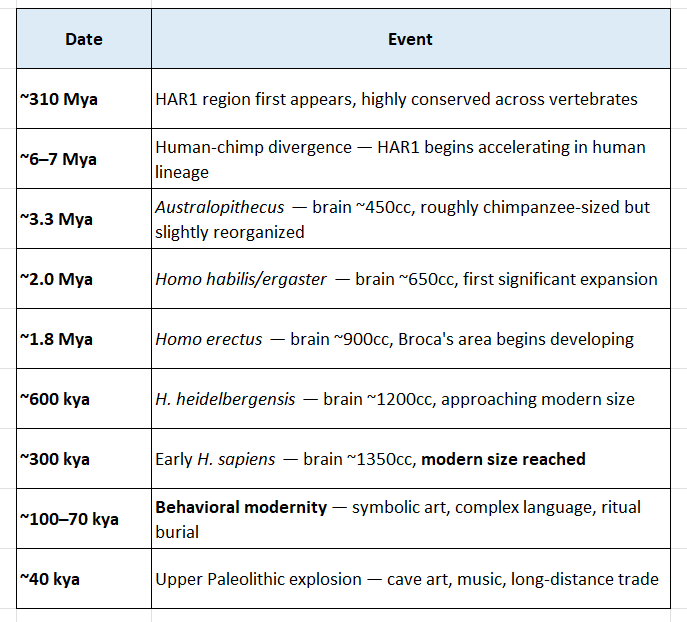
Notice the critical paradox: modern brain size is reached at ~300,000 years ago, but behavioral modernity only erupts at ~100,000–40,000 years ago. This suggests the decisive changes were not in volume but in internal wiring and connectivity — exactly where HARs operate.
4. ARHGAP11B — The Cortical Folding Gene
~3.5 million years ago — a uniquely human gene duplication event created ARHGAP11B, which does not exist in chimpanzees or other great apes.
This gene causes basal radial glia (stem cells in the developing cortex) to proliferate far more than in other primates. The result: cortical folding — the characteristic wrinkled surface of the human brain.
Experiments published in 2015 (Stahl et al., Science) showed that inserting ARHGAP11B into mouse and ferret embryos caused their normally smooth cortices to fold, producing a gyrification pattern resembling the human brain. This was a landmark moment — a single gene duplication, ~3.5 million years old, demonstrably reshaping cortical architecture.
5. NOTCH2NL — The Great Delay
2018 — Two simultaneous papers in Cell (Fiddes et al., Suzuki et al.) identified NOTCH2NL genes — another uniquely human duplication, originating approximately 3–4 million years ago (with the functional version appearing ~1 million years ago).
NOTCH2NL delays the differentiation of cortical progenitor cells — meaning they divide more times before becoming neurons. More divisions = more neurons = larger cortex.
Crucially, deletions of NOTCH2NL in modern humans are associated with microcephaly and autism. Duplications are associated with macrocephaly and schizophrenia. The gene sits at a dosage sweet spot — suggesting the human brain expanded to near the edge of developmental stability.
6. The 2,700 HARs — The Full Landscape
After Pollard’s original 202, subsequent analyses expanded the catalog:

7. The FOXP2 Connection — Language
~2.5 million years ago — two specific mutations in the FOXP2 gene — the so-called “language gene” — occurred uniquely in the human lineage. Neanderthals share these mutations, suggesting they appeared before the human-Neanderthal split (~500–700 kya) and that Neanderthals may have had some capacity for structured vocalization.
FOXP2 is not a “language gene” per se — it is a transcription factor that regulates dozens of genes involved in the fine motor control of the vocal tract and the basal ganglia-cortical circuits underlying procedural learning and speech.
8. The Deepest Mystery — The Timing Gap
Here is the unresolved tension that makes this field so alive:
Most HAR mutations and uniquely human gene duplications accumulated 3–6 million years ago
Yet behavioral modernity — language, symbolic thought, art — only explodes ~100,000–50,000 years ago
This is a gap of 3 million years between the genetic changes and their full behavioral expression.
Leading hypotheses:
Threshold model — the changes accumulated silently until a critical network complexity was reached
Cultural catalyst — the genes created the potential, but a cultural innovation (proto-language, social structure) was needed to unlock it
Epigenetic regulation — the HAR regions changed gene regulation patterns that only manifest under specific developmental or social conditions
Missing mutations — the decisive changes are more recent and still being identified
The genome is beginning to look less like a blueprint and more like a deep historical archive — where the story of human cognition is written in the language of accelerated mutations, gene duplications, and regulatory switches that were set in motion millions of years before the first cave painting.
Here is the full pipeline — from tissue sample to the decoding of what makes us human, step by step.
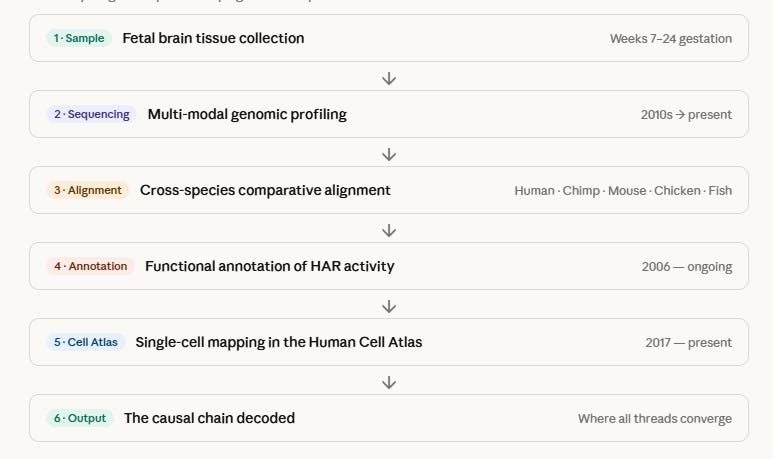
The six stages form a closed loop — from a needle of tissue taken during week 12 of gestation, all the way to understanding why a mutation that happened 6 million years ago built the human neocortex.
The most important stage is arguably stage 5 — the single-cell resolution. Before scRNA-seq, researchers knew HARs were active “in the brain.” After it, they knew HARs are active in this specific cell type, in this specific week, in this specific cortical layer. That precision is what turns correlation into mechanism.
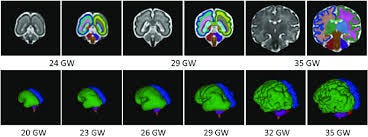

The Ventricular Zone — Where the Human Brain Builds Itself
This is one of the most precise and important questions in all of developmental neuroscience. The ventricular zone is not just a location for HAR1 activity — it is the foundational germinal layer of the entire cortex, and understanding why HAR1 is active there unlocks the whole story of how the human neocortex became what it is.
What the Ventricular Zone Is
The ventricular zone (VZ) is a thin layer of cells lining the inner surface of the developing brain — surrounding the fluid-filled ventricles. It exists from approximately gestational week 5 through week 20, and it is the birthplace of virtually every neuron in the cerebral cortex.
It is packed with radial glia — the master stem cells of the brain. These cells do two things simultaneously: they divide to produce neurons, and they extend long fibres outward like scaffolding, along which newborn neurons climb to reach their correct layer in the cortex.
Why HAR1 Is Specifically Active There
HAR1 does not encode a protein. It encodes a non-coding RNA — HAR1F — whose sole function is structural: it folds into a precise three-dimensional shape that interacts with other molecules. The shape matters as much as the sequence.
HAR1F is expressed specifically in Cajal-Retzius neurons, which are born in the ventricular zone during weeks 7–16 and then migrate to the outermost layer of the cortex — the marginal zone. From there, they secrete a protein called Reelin.
Reelin is the master organiser of cortical lamination — the six-layer architecture of the human neocortex. It acts as a positional signal telling incoming neurons where to stop migrating. Without Reelin, cortical layers collapse and merge into a disordered mass. This condition — called lissencephaly — produces a smooth, unfolded brain with severe cognitive impairment.
So the chain is:
HAR1F RNA (expressed in Cajal-Retzius neurons born in VZ) → correct RNA folding → appropriate interaction with transcription factor CTCF → proper Cajal-Retzius neuron development → Reelin secretion → 6-layer cortical architecture → the physical substrate of human cognition.
The Human-Specific Difference
The 18 human-specific mutations in HAR1 change the secondary structure of the HAR1F RNA molecule — the way it folds. The ancestral (chimpanzee) version folds into a different shape. This shape difference alters how the RNA interacts with CTCF, which in turn affects how many Cajal-Retzius neurons are produced and how efficiently they secrete Reelin.
The ventricular zone is where this entire cascade is initiated. It is the first domino — and it falls during a remarkably narrow window: weeks 7 through 16. Outside this window, HAR1F is essentially silent.
Why the VZ and Not Later Stages?
Cortical development follows a strict inside-out sequence: the deepest layers (VI, V) are built first, the superficial layers (II, III) last. Each layer requires the previous one to be correctly established. If the Cajal-Retzius neurons in the VZ do not set up the Reelin gradient correctly at week 8, every subsequent layer of the cortex is displaced — like building a tower on a crooked foundation.
This is why HAR1 activity in the VZ is so consequential: a small change in RNA folding in a small population of cells during a narrow developmental window propagates into the entire macro-architecture of the human brain. The VZ is the point of maximum leverage in cortical development — and HAR1 sits precisely at that fulcrum.
In evolutionary terms, this is elegant and efficient: nature did not need to rebuild the brain from scratch. It needed only to remodel a single RNA molecule in the right cells at the right moment — and the result, accumulated over 6 million years, was the human neocortex.

ATAC-seq — Reading the Open Genome
To understand ATAC-seq, you first need to understand the fundamental problem it solves. The human genome contains 3 billion base pairs of DNA — but the vast majority of it is physically inaccessible at any given moment in any given cell. It is wound tightly around protein spools called nucleosomes, compacted into a dense structure called chromatin. A gene enhancer that is buried inside chromatin cannot function — the transcription machinery simply cannot reach it.
The question ATAC-seq answers is: which regions of the genome are physically open right now, in this cell type, at this moment?
Those open regions are, almost by definition, the ones that are doing something — regulatory elements, active enhancers, promoters. Closed regions are silent. ATAC-seq is therefore not a sequence reader — it is a topological map of regulatory activity.
The deepest insight here is philosophical as much as technical. ATAC-seq does not read the genome as a text — it reads it as a physical state. The same DNA sequence, in two different cell types or two different species, can be completely open in one and completely closed in the other. The sequence did not change. What changed is which proteins are sitting on it, which marks are written on the histones, which regulatory circuits are active in that cell’s developmental history.
This is why the human-chimpanzee comparison is so powerful when done with ATAC-seq. The HAR1 sequence in a chimpanzee is only 18 bases different from the human version. But those 18 bases change the three-dimensional folding of the RNA — which changes how the enhancer is bound by regulatory proteins — which determines whether the chromatin is open or closed in that cell type at that developmental moment. ATAC-seq makes that difference visible as a peak that is present in human oRG cells and absent in the equivalent chimpanzee cells. A 6-million-year evolutionary story, compressed into a single accessibility peak on a sequencing track.

PhyloP — The Statistical Engine Behind HAR Discovery
PhyloP is one of the most elegant algorithms in comparative genomics. At its core, it answers a deceptively simple question: given how slowly this DNA region has changed across 300 million years of vertebrate evolution, how improbably fast did it change in the human lineage? The answer is expressed as a single number — the PhyloP score — and when that number is extreme enough, the region becomes a HAR.
The algorithm operates at the intersection of evolutionary biology, molecular phylogenetics, and statistical hypothesis testing. Let us build it up from its foundations.
There is one deeper subtlety worth holding in mind. PhyloP is a frequentist test — it asks how improbable the observed data is under the null hypothesis of neutrality. But it cannot by itself distinguish between three very different biological causes of acceleration:
Natural selection — some change in the environment or developmental context made the new sequence beneficial, and it was positively selected across the population. This is the most dramatic interpretation and likely explains a significant portion of HAR accelerations.
Biased gene conversion (BGC) — during meiotic recombination, GC-ending sequences are preferentially incorporated over AT-ending ones at conversion hotspots. Because many HAR substitutions happen to be AT→GC changes, some researchers argue BGC could mimic selection without any fitness advantage. This is genuinely contested for some HARs.
Relaxed constraint — if the region lost its ancestral function in the human lineage, it could evolve neutrally while the rest of the genome is still conserved, creating a spurious acceleration signal. This seems unlikely for HAR1 given its demonstrable activity in Cajal-Retzius neurons, but it remains a theoretical alternative for less well-studied HARs.
The PhyloP score is thus the beginning of the inquiry, not the end. It points the finger with extraordinary statistical precision — and then the organoids, the ATAC-seq, the base editing experiments do the work of understanding why those bases changed, and what they do.

H3K27ac — The Molecular Signature of an Active Enhancer
H3K27ac is a post-translational modification of a histone protein. To understand why it marks active enhancers — and why its presence at HAR regions is so significant — we need to descend through four levels of biology: from the nucleosome, to the histone tail, to the acetyl group, to the regulatory machinery that reads it.
Work through all five tabs — the story builds from physical chemistry at the atomic level all the way up to genome-wide detection of HAR enhancer activity.
There is something philosophically arresting in all of this. The 18 mutations in HAR1 that accumulated over 6 million years did not build a new protein. They did not create a new gene. They remodelled the three-dimensional shape of a small non-coding RNA — which changed how CBP/p300 was recruited to that locus — which determined whether a lysine at position 27 on histone H3 received an acetyl group — which neutralised a positive charge — which loosened the grip of a nucleosome on 147 base pairs of DNA — which allowed a transcription factor to bind — which activated the Cajal-Retzius neurons — which secreted Reelin — which built the six-layer human neocortex.
The entire architecture of human cognition is downstream of a charge neutralisation on a single amino acid, written by an enzyme whose activity was redirected by an RNA molecule that changed shape over millions of years of selection.
This is what molecular biology, at its deepest, reveals: the universe of a single chemical bond.
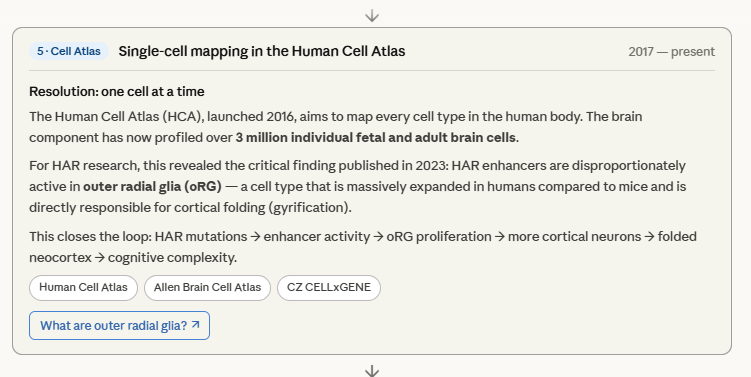
Outer Radial Glia — The Cell That Built the Human Mind
Of all the discoveries to emerge from comparative neuroscience in the last two decades, the outer radial glia may be the most consequential. It is a single cell type — absent in mice, rudimentary in most primates, massively expanded in humans — that is largely responsible for the extraordinary size, folding, and cognitive capacity of the human neocortex. Understanding it means understanding the cellular engine of human intelligence.
There is something that deserves particular reflection — the species comparison. The mouse has essentially no oSVZ and essentially no oRG. Its cortex is smooth and its neuron count is roughly 14 million. The human oSVZ is so thick during peak neurogenesis that it becomes the dominant germinal zone, generating the majority of cortical neurons — 16 billion in the finished cortex. And yet the genetic distance between a mouse and a human is not proportionally greater than the functional distance between their brains. The oRG expansion was not a gradual scaling up — it was a threshold transition, triggered by a small number of gene duplications, that crossed from one qualitative regime of cortical organisation into another.
ARHGAP11B, which is perhaps the single most important gene in this story, did not evolve from scratch. It is a truncated copy of an existing gene, created by a retrotransposition event 3.5 million years ago — probably in an early Australopithecus or earlier hominin — with a reading frameshift that accidentally created a new functional protein. Evolution did not design the human brain. It stumbled upon a duplication, retained it because its effects were beneficial, and 3.5 million years later that accident became the cellular engine of human consciousness.
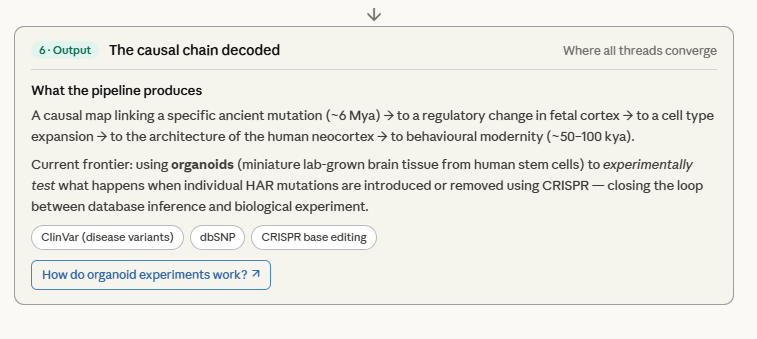
Brain Organoids as Time Machines for HAR Mutations
This is the sharpest experimental frontier in all of human evolutionary genomics. For the first time in history, it is possible to take a specific mutation that occurred 6 million years ago in an ancestral hominin, introduce it into living human neural tissue, and watch what happens to the developing cortex in real time. The organoid is not a metaphor — it is a functional laboratory for testing the causal claims of evolutionary genomics.
The story moves from the logical necessity of the organoid system, through the four experimental strategies, the published timeline of key experiments, the full measurement toolkit, and finally the frontier and limits.
The timeline on tab 3 deserves particular attention as an intellectual arc. The field moved in a decade from ARHGAP11B inserted into a mouse (2015) — a blunt demonstration that a single gene can fold a cortex — to base-resolution causal mapping of individual HAR nucleotides in human tissue (2022). Each step sharpened the resolution: from gene to enhancer, from enhancer to specific base, from tissue to single cell, from correlation to dose-dependent causation.
What strikes me most deeply about the entire organoid approach is its philosophical reversal of the usual direction of evolutionary inference. Normally we read evolution backward — we look at what exists today and infer what must have happened in the past. The organoid experiment runs the clock forward: we take a cell carrying an ancestral sequence that existed 6 million years ago, grow it into living cortical tissue, and watch the past literally develop in a dish. The Australopithecine cortex is not entirely beyond reach — it can be partially reconstructed, tested, and compared to the human cortex that evolved from it.
The limit that tab 5 ends on is the one that most deserves continued reflection. The organoid demonstrates the cellular hardware. The 3-million-year gap between the genetic changes and behavioural modernity remains unexplained by any experiment so far. That gap is the most important open question in the biology of human consciousness — and no organoid, however sophisticated, can yet touch it.